Ancient DNA laboratory work
 Ancient DNA (aDNA) is typically extracted from individuals long-since dead, from sources including bones, teeth, shells, coprolites, and others. Ancient DNA has physical and chemical properties unlike DNA from still living cells. Following death, DNA is witness to degradation and chemical modification from nucleases, microorganisms, hydrolysis, UV radiation, and oxidation. When these forces fail to completely destroy the DNA, aged and degraded specimens may retain low copy number molecules that are short with regards to strand length and typically exhibit miscoding lesions which can appear as “mutations” when, in fact, they are artifacts. Using aDNA methods involves paying close attention to contaminating exogenous DNA that can originate from modern and ancient sources, and the co-extraction of PCR inhibitors (read more about aDNA at the Kemp Lab site). As part of the Kemp Lab, I have extracted and analyzed ancient DNA from a wide range of taxa, including human rib bones from pre-contact Native Americans, and northern fur seals rib bones dating to 3500 years before present. I also have collaborated in small projects by extracting ancient DNA from rodent teeth, coprolites from ancient Pueblo Native Americans, and plant material from lake sediment. I have recently collaborated with mollusk biologists from The University of Idaho to develop a purification method capable of extracting high quality DNA from empty mollusk shells collected in the field from species which are now thought to be extinct.
Ancient DNA (aDNA) is typically extracted from individuals long-since dead, from sources including bones, teeth, shells, coprolites, and others. Ancient DNA has physical and chemical properties unlike DNA from still living cells. Following death, DNA is witness to degradation and chemical modification from nucleases, microorganisms, hydrolysis, UV radiation, and oxidation. When these forces fail to completely destroy the DNA, aged and degraded specimens may retain low copy number molecules that are short with regards to strand length and typically exhibit miscoding lesions which can appear as “mutations” when, in fact, they are artifacts. Using aDNA methods involves paying close attention to contaminating exogenous DNA that can originate from modern and ancient sources, and the co-extraction of PCR inhibitors (read more about aDNA at the Kemp Lab site). As part of the Kemp Lab, I have extracted and analyzed ancient DNA from a wide range of taxa, including human rib bones from pre-contact Native Americans, and northern fur seals rib bones dating to 3500 years before present. I also have collaborated in small projects by extracting ancient DNA from rodent teeth, coprolites from ancient Pueblo Native Americans, and plant material from lake sediment. I have recently collaborated with mollusk biologists from The University of Idaho to develop a purification method capable of extracting high quality DNA from empty mollusk shells collected in the field from species which are now thought to be extinct.
Bayesian Tree Sampling Analysis
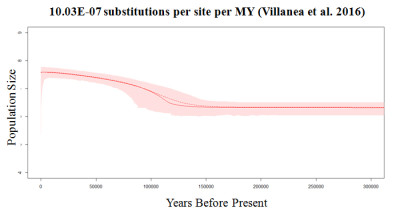 In 2007, Alexei J Drummond and Andrew Rambaut published BEAST: Bayesian evolutionary analysis by sampling trees, a program which utilizes a neutral coalescent framework to relate individuals – as represented by sequence data – backwards in time in a genealogy. BEAST incorporates Bayesian MCMC analysis by allowing for the information provided by both the sequence data and user input (a set of priors) to jointly estimate an array of genealogies. Together, these genealogies represent a sample of the likely possibilities of coalescence back in time to the individuals’ most recent common ancestor (MRCA), under a given demographic model. These genealogies can be represented topographically as trees, thus the method is popularly referred to as tree sampling analysis. As part of my research, I have used BEAST to explore the demographic history of northern fur seals, utilizing a mix of ancient and modern DNA to reconstruct three population expansions which occurred in the last ~70,000 years. This form of analysis involved the exploration of various demographic models, including Bayesian Skyline plots. Ongoing research focuses on the estimation of accurate molecular rates, for which simulation work is invaluable in evaluating the accuracy and sensitivity of estimates made using BEAST from aDNA sequence data.
In 2007, Alexei J Drummond and Andrew Rambaut published BEAST: Bayesian evolutionary analysis by sampling trees, a program which utilizes a neutral coalescent framework to relate individuals – as represented by sequence data – backwards in time in a genealogy. BEAST incorporates Bayesian MCMC analysis by allowing for the information provided by both the sequence data and user input (a set of priors) to jointly estimate an array of genealogies. Together, these genealogies represent a sample of the likely possibilities of coalescence back in time to the individuals’ most recent common ancestor (MRCA), under a given demographic model. These genealogies can be represented topographically as trees, thus the method is popularly referred to as tree sampling analysis. As part of my research, I have used BEAST to explore the demographic history of northern fur seals, utilizing a mix of ancient and modern DNA to reconstruct three population expansions which occurred in the last ~70,000 years. This form of analysis involved the exploration of various demographic models, including Bayesian Skyline plots. Ongoing research focuses on the estimation of accurate molecular rates, for which simulation work is invaluable in evaluating the accuracy and sensitivity of estimates made using BEAST from aDNA sequence data.
Approximate Bayesian Computation
 As an alternative when dealing with inconclusive Bayesian tree sampling analyses, a model fitting approach called approximate Bayesian computation (ABC) has become widely adopted for its ability to infer complex models of demographic evolution applicable to small empirical sample sets. ABC is a likelihood-free method; it avoids calculating the likelihood portion of Bayesian inference. The intuition behind it is that simulated data under a known random process produce distributions of parameters which are proportional to their likelihood, and thus proportional to their posterior probability. Under ABC, an approximation of the likelihoods is generated by comparing simulated data against empirical data. In ABC applied to genetic studies of demography, complete genealogies are simulated computationally forward in time to produce alignments of sequences, using the coalescent model as a foundation. To compliment my research on the demographic history of northern fur seals, I have used approximate Bayesian computation to describe two population events occurring in the Holocene period, for which BEAST analyses lacked the sensitivity to capture. As an additional benefit, simulated data can be used in to test the accuracy of specific ABC analyses applied to aDNA data. We explored the statistical power of utilizing aDNA mitochondrial sequence data of various sized ranging from small <150 base pair fragments to the full coding region of the mitochondrial genome, and found that the addition of aDNA to ABC analyses does increase statistical power over analysis using modern DNA alone, however any additional benefits of including aDNA information decreases asymptotically, as longer sequences reveal almost as much information as short sequences, a consequence of mitochondrial DNA representing a single linked locus.
As an alternative when dealing with inconclusive Bayesian tree sampling analyses, a model fitting approach called approximate Bayesian computation (ABC) has become widely adopted for its ability to infer complex models of demographic evolution applicable to small empirical sample sets. ABC is a likelihood-free method; it avoids calculating the likelihood portion of Bayesian inference. The intuition behind it is that simulated data under a known random process produce distributions of parameters which are proportional to their likelihood, and thus proportional to their posterior probability. Under ABC, an approximation of the likelihoods is generated by comparing simulated data against empirical data. In ABC applied to genetic studies of demography, complete genealogies are simulated computationally forward in time to produce alignments of sequences, using the coalescent model as a foundation. To compliment my research on the demographic history of northern fur seals, I have used approximate Bayesian computation to describe two population events occurring in the Holocene period, for which BEAST analyses lacked the sensitivity to capture. As an additional benefit, simulated data can be used in to test the accuracy of specific ABC analyses applied to aDNA data. We explored the statistical power of utilizing aDNA mitochondrial sequence data of various sized ranging from small <150 base pair fragments to the full coding region of the mitochondrial genome, and found that the addition of aDNA to ABC analyses does increase statistical power over analysis using modern DNA alone, however any additional benefits of including aDNA information decreases asymptotically, as longer sequences reveal almost as much information as short sequences, a consequence of mitochondrial DNA representing a single linked locus.
Analytical Modeling
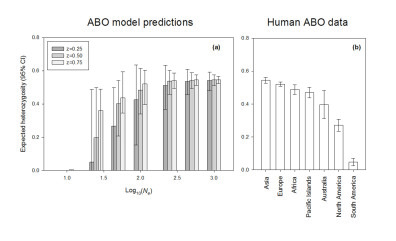 The simulation of biological processes through stochastic or deterministic models represents an invaluable tool to explore evolutionary processes, particularly those based on the predictions of population genetics. Mathematical models can generate results easily comparable with empirical genetic data, as a heuristic tool to understand the biological processes which shape genetic polymorphism. I am interested in such heuristic approaches, and have applied them in the past to study the interaction of balancing selection and genetic drift in modern and ancient populations. Long-term balancing selection is rarely observed in nature, as there few examples of the maintenance of polymorphism over evolutionary times. The best known examples are S-alleles for incompatibility in flowering plants and fungi, the major incompatibility complex (MHC) in vertebrates, and the human ABO blood groups locus. For the ABO locus, empirical population data is suggestive of the influence of balancing selection; however the physiological mechanism remains unknown. A mathematical model incorporating balancing selection and genetic drift which simulated the maintenance and loss of ABO polymorphism in human populations produces patterns comparable to living populations today, supporting the role of balancing selection, even in the absence of a physiological mechanism. Furthermore, the model predicted that the low observed polymorphism in the ABO locus in Native American populations was a consequence of serial bottlenecks, which reduced the effective population size of the American founder populations below numbers at which balancing selection would operate efficiently.
The simulation of biological processes through stochastic or deterministic models represents an invaluable tool to explore evolutionary processes, particularly those based on the predictions of population genetics. Mathematical models can generate results easily comparable with empirical genetic data, as a heuristic tool to understand the biological processes which shape genetic polymorphism. I am interested in such heuristic approaches, and have applied them in the past to study the interaction of balancing selection and genetic drift in modern and ancient populations. Long-term balancing selection is rarely observed in nature, as there few examples of the maintenance of polymorphism over evolutionary times. The best known examples are S-alleles for incompatibility in flowering plants and fungi, the major incompatibility complex (MHC) in vertebrates, and the human ABO blood groups locus. For the ABO locus, empirical population data is suggestive of the influence of balancing selection; however the physiological mechanism remains unknown. A mathematical model incorporating balancing selection and genetic drift which simulated the maintenance and loss of ABO polymorphism in human populations produces patterns comparable to living populations today, supporting the role of balancing selection, even in the absence of a physiological mechanism. Furthermore, the model predicted that the low observed polymorphism in the ABO locus in Native American populations was a consequence of serial bottlenecks, which reduced the effective population size of the American founder populations below numbers at which balancing selection would operate efficiently.
Coalescent-based admixture simulations
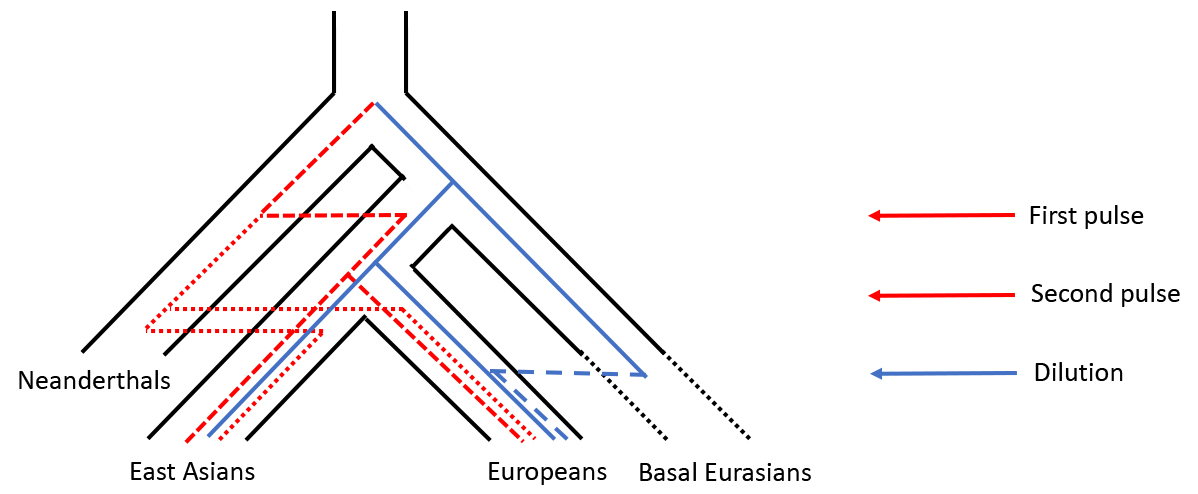 The neutral coalescent framework allows for the study of population admixture, by looking at a collective of genealogies with various architectures. Each genealogy represents a possible relation between individuals, the abundance of a genealogy connecting two individuals from different populations representing both the probability and the frequency of admixture. Simulations under this framework are easy to understand, as the model has simple expectations. I have used this approach to study the contribution of neanderthals into the genomes of modern humans. Neanderthals and humans overlapped geographically for a period of over 30,000 years following human migration out of Africa. During this period, Neanderthals and humans interbred, as evidenced by Neanderthal portions of the genome carried by non-African individuals today. A key observation is that the proportion of Neanderthal ancestry is ~20% higher in East Asian individuals relative to European individuals. I explored various demographic models that could explain this observation. These include distinguishing between a single admixture event and multiple Neanderthal contributions to either population, and the hypothesis that reduced Neanderthal ancestry in modern Europeans resulted from more recent admixture with a ghost population that lacked a Neanderthal ancestry component (the “dilution” hypothesis). Using machine learning, the output from the simulations can compared to genomic data from the 1000 genomes project. This method was able to distinguish between various demographic models, and found that of the complex models, a model of multiple episodes of gene flow into both East Asian and European populations fits the data best. These findings suggest long-term, complex interaction between humans and Neanderthals.
The neutral coalescent framework allows for the study of population admixture, by looking at a collective of genealogies with various architectures. Each genealogy represents a possible relation between individuals, the abundance of a genealogy connecting two individuals from different populations representing both the probability and the frequency of admixture. Simulations under this framework are easy to understand, as the model has simple expectations. I have used this approach to study the contribution of neanderthals into the genomes of modern humans. Neanderthals and humans overlapped geographically for a period of over 30,000 years following human migration out of Africa. During this period, Neanderthals and humans interbred, as evidenced by Neanderthal portions of the genome carried by non-African individuals today. A key observation is that the proportion of Neanderthal ancestry is ~20% higher in East Asian individuals relative to European individuals. I explored various demographic models that could explain this observation. These include distinguishing between a single admixture event and multiple Neanderthal contributions to either population, and the hypothesis that reduced Neanderthal ancestry in modern Europeans resulted from more recent admixture with a ghost population that lacked a Neanderthal ancestry component (the “dilution” hypothesis). Using machine learning, the output from the simulations can compared to genomic data from the 1000 genomes project. This method was able to distinguish between various demographic models, and found that of the complex models, a model of multiple episodes of gene flow into both East Asian and European populations fits the data best. These findings suggest long-term, complex interaction between humans and Neanderthals.
Trait-specific speciation rates
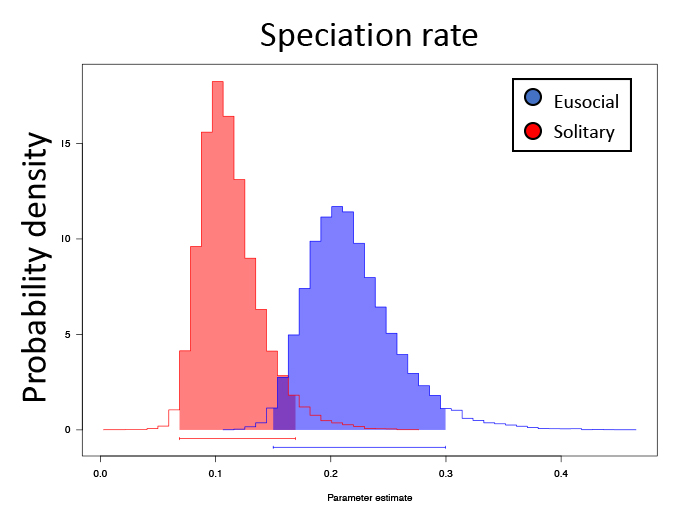 State Speciation and Extinction (SSE) methods allow us to observe if a species is evolving at a different rate than it’s related taxa, but more importantly allows us to predict if a trait is responsible for driving the differential evolution rate between species. Halictid bees, or ‘sweat’ bees, are an ideal model for studying the transitions between solitary and eusocial reproduction. Eusociality has been gained and lost repeatedly in this group, and as a result, closely-related halictid species span the full spectrum of social forms, from solitary to eusocial. I am interested in studying if the transition from solitary to eusocial reproduction, or the converse transition from eusocial to solitary reproduction, has an effect on genetic diversity, population structure, and in particular, speciation rates. I tested if eusociality is driving state-dependent diversification rates using State Speciation and Extinction (SSE) methods across the larger phylogeny of Halictidae. The phylogenetic comparisons indicate that eusociality is associated with higher rates of speciation than solitary reproduction, a result that is is robust across likelihood-based and non-parametric SSE tests as well as hidden state models.
State Speciation and Extinction (SSE) methods allow us to observe if a species is evolving at a different rate than it’s related taxa, but more importantly allows us to predict if a trait is responsible for driving the differential evolution rate between species. Halictid bees, or ‘sweat’ bees, are an ideal model for studying the transitions between solitary and eusocial reproduction. Eusociality has been gained and lost repeatedly in this group, and as a result, closely-related halictid species span the full spectrum of social forms, from solitary to eusocial. I am interested in studying if the transition from solitary to eusocial reproduction, or the converse transition from eusocial to solitary reproduction, has an effect on genetic diversity, population structure, and in particular, speciation rates. I tested if eusociality is driving state-dependent diversification rates using State Speciation and Extinction (SSE) methods across the larger phylogeny of Halictidae. The phylogenetic comparisons indicate that eusociality is associated with higher rates of speciation than solitary reproduction, a result that is is robust across likelihood-based and non-parametric SSE tests as well as hidden state models.